So to recap from yesterday...we planned our cell viability assay to test whether the Type II Diabetes drug Metformin can reduce cell viability of three cell lines - MCF10A, MCF12A and MCF7.
Today! We will start our experiment but plating our cells into the 96-well plate to treat. Day 2: Plating Our Experiment It's important to remember our information we had yesterday (reagents, equipment, plate plan). Part 1: Making A Cell Suspension As I've explained before the cell lines are grown in a flask and attach to the plastic. But we want them to detach to be able to plate them up (don't worry the majority will survive not detached for a short time). This is called a cell suspension. We create a cell suspension by first removing the media. This gets rid of all the dead cells. The media also inhibits the reagent we'll use to detach the cells so we need to get rid of it. Once the media is removed we wash the flask with PBS to make sure that there is NO media left. PBS (or phosphate buffered saline to those interested) is a clear liquid that does not react with other liquids or cells and is often used for washing cells etc. (like soap but less harsh - also we don't use soap on cells...). Once the cells are nice and clean we add EDTA/Trypsin. Basically these chemicals together "scrape" the cells off the bottom of the flask (for more in depth explanation see yesterday's post). Depending on the cell line you use this can take 2 minutes (MCF7) or 15 minutes (MCF10A and MCF12A). After you leave it for it's needed time all the cells detach off the plate, leaving you with a cloudy liquid chock-full of cells (see picture 2). BUT EDTA/Trypsin is toxic to cells and will eventually kill them so you need to neutralize it. Remember when I talked about media and what it does to EDTA/Trypsin? You got it. We add media to the cell suspension to neutralize the EDTA/Trypsin. We then put the cell suspension in a 50ml falcon (see picture 3). As you can see it's all cloudy because of all the cells floating around. We could use these cells however there is a chance that not all of the EDTA/Trypsin is neutralized so we want to get rid of the liquid and replace it with new liquid while keeping our cells. We do this by first centrifuging the cells at a very low speed. The centrifuge will force all of the cells to fall to the bottom of the flask because they are "heavier" than the liquid (see picture 4). We can then take off the liquid while leaving the cells intact and add new media (free of toxins). This puts the cells back to floating around (like picture 3) and there we have our cell suspension.
Part 2: Counting Our Cells
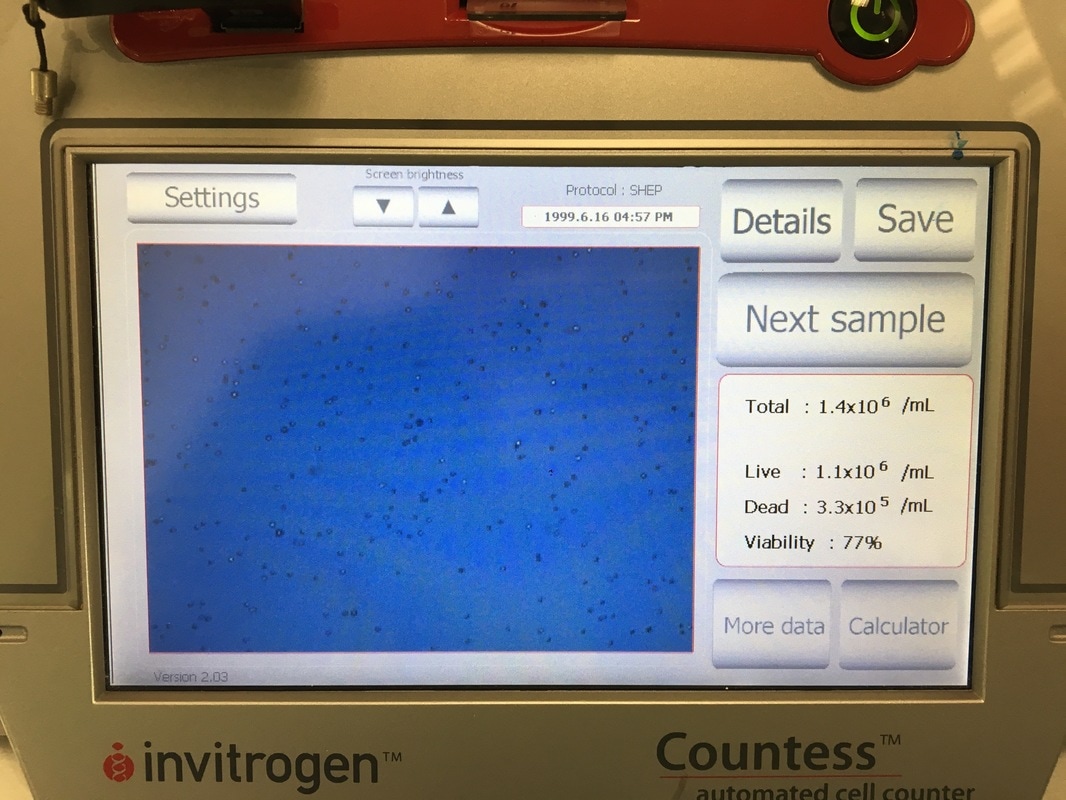
It is important in an experiment to have a similar number of cells in each cell line at the END of the experiment so we can make comparisons. As I said before, these cell lines all grow at a similar pace so plating the same number of cells should give us roughly the same number of cells at the end of our experiment. Now we're hypothesising that Metformin will "kill" some of these cells but it may do this better in some cell lines compared to others. So how do you know you have the same amount of cells at the end? We add a control. The control is 0mM on our plan. This basically has no drug added to it, meaning they should grow without dying (too much) and will represent the total number of cells alive (or 100% alive cells). So on to counting. Cells are counted by adding a blue dye (not crystal violet) but another dye, Trypan Blue. Trypan blue ONLY stains cells that are dead. So we can distinguish between alive (white) and dead cells (blue). Now you can do this manually under a microscope OR you can do it the way I do it which is with a machine! The machine measures how much blue and how much white there is an calculates how many alive cells there are in 1ml of your cell suspension. To do this we take 20ul (which is microlitres or 0.002ml) of trypan blue and add it to 20ul of cell suspension. We then put it in a little cartridge (picture 5) and place it into the machine. The machine then counts the cells and tells us how many we have (picture 6 - total). It tells you how many cells are dead, how many are alive and how good your sample is (viability %). I use the total value because I find that when I use the "Live" value my plates are always over confluent at the end of the experiment (i.e. there's too many cells and not enough space so the cells start to die). But that's me. When I use the "Total" value the controls all have very similar cell numbers at the end so I'm happy with that.
Now to calculate how much cell suspension we need and how much media we need (picture 7). First we convert our "Have" value to the same number are our "Want" value (i.e. make sure the "Have" value is x105). In the case of MCF7 we don't need to do that but for MCF10A and MCF12A we do. So we take one 10 away from x106 and multiply the number before the x by 10. We then divide WANT/HAVE. This gives a value in ml telling us how much cell suspension we need (Value in RED). We then take that away from the "Total Volume" to get how much media we need (value in BLUE).
Part 4: Plating Our Experiment (YAY)
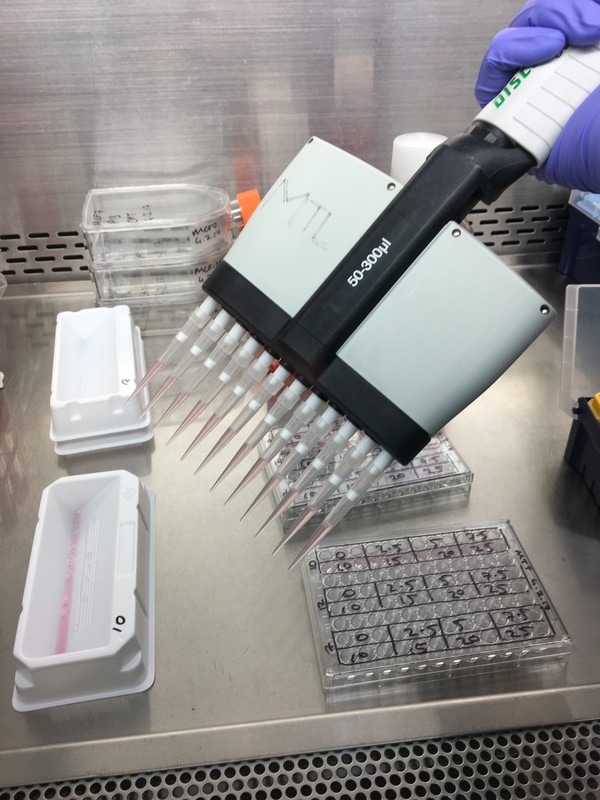
We now have everything we need to plate. We have our plating plan and we have the volumes of cell suspension and media. The final thing we do before we start is draw our plate plan onto the lid of our plate just so we don't forget which well is which. We also label the cell lines and add our name, date and experiment (picture 8). To plate our cells we add the final volume of media to the final volume of cell suspension in a labelled 50ml falcon. We next use a multichannel pipette to add the cells to the plate (see pictures 9 and 10). This is for three reasons: (1) this ensures equal distribution of cells, (2) it's faster then doing each well individually and (3) I have a wrist injury so using a multichannel pipette makes life easier. The multichannel takes up 0.1ml of cell suspension from a reservoir into each pipette tip and adds it evenly to each well. Once all the wells you want to be plated are full of your chosen cell line you then leave the plate in an incubator at 37°C and 5% CO2 overnight.
And that's it! We have now plated our experiment!!!
Tomorrow we will be treating our cells! So exciting!! NOTE: Apologies for the late posting, I was at a conference today and had no time to write this up.
0 Comments
Leave a Reply. |
AuthorMy name is Caitriona and I am a PhD student at Imperial College London, UK. Categories
All
|