|
Recently the European Association for Cancer Research (EACR), who as you know funded me to travel to IARC in Lyon to work, launched a blog competition. The idea of the competition was to write a short blog-style post about "Life in the Lab". You were allowed to submit as many blogs as you liked so I submitted three:
I found out today that the first one (Engaging with the (scary) public – why we need to do it and do it now) was shortlisted as one of the best blogs submitted. So while I didn't win, it will still get published in the next few months. BUT what I also learnt was that they liked my other two so much that they are going to published anyway probably early next year! How awesome is that! Since they are going to be published I won't be adding them up here but watch this space. Once they become available on the EACR magazine website I will let you all know! 
0 Comments
Yesterday I was at the New Scientist Live exhibition in the ExCEL London with CRUK (sorry I am terrible at taking photos so eh no evidence). My job there was to chat to the future scientists from 13 year olds to 23 year olds about how to get a career in research and what it was like to do a PhD. And my one biggest piece of advice was do coding! Bioinformatics is the future. Seriously, even if you hate maths (like me) learning some code language will help you in anything you do.
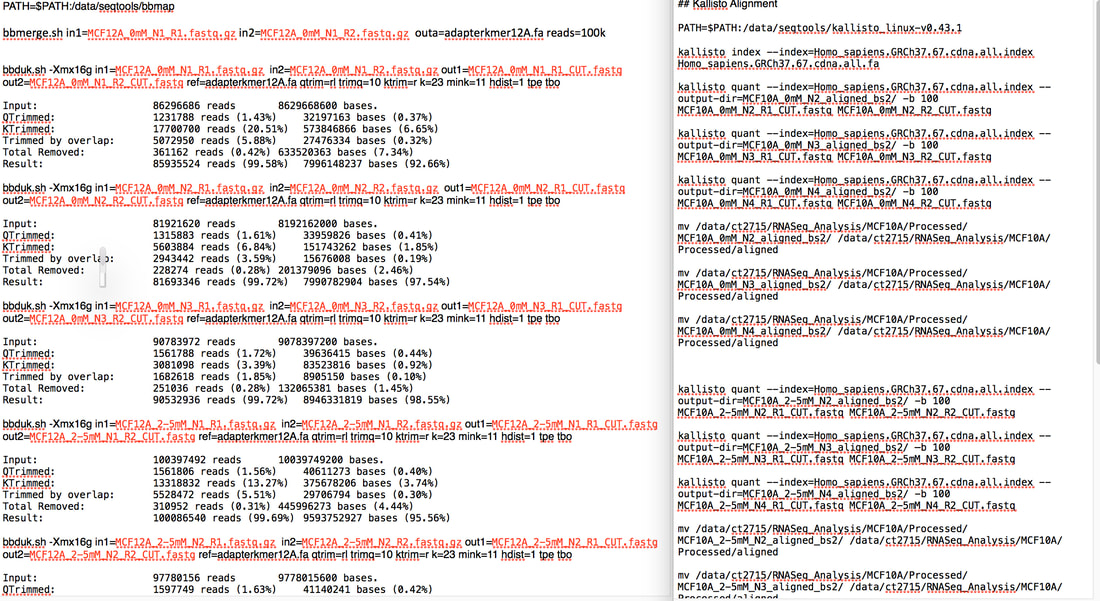
My other piece of advice was that if you want a PhD google www.findaphd.com/ because it does exactly what it says on the tin. It found me a PhD. So what is bioinformatics and why is it important? Bioinformatics is basically the analysis of biological data. Biological data usually means sequencing data. You use a coding language such as R, python or unix to analyse this data. The above picture is my code for doing quality control on my RNA sequencing data using unix. I can also (barely) use R. To me python is a snake... Bioinformatics is important because we can take massive amounts of data from hundreds or thousands of different samples and use this to generate more knowledge about areas such as cancer. This can be sequencing the entire compliment of genes in tumours of hundreds of patients to see if there are common mutations. This can tell us what these tumours have in common and what they don't. You can use this information for example to find "driver mutations" which are mutations commonly found in early stage tumours (p53 mutation is a driver mutation as some cancers require loss of p53 to develop into cancer). The idea of using whole genome sequencing in cancer has come up a lot recently in the news. And the people who are taking this data and finding interesting results are bioinformaticians. Pretty much every cancer research lab in the world is generating data which needs to be analysed computationally. The data you get collectively from sequencing etc. is called "Big Data". However while most labs are generating this data, a large amount of those labs have no one to analyse it. We need more bioinformaticians to take this data and make it useful. Hence why bioinformatics is the future. The dawn of the era of joint experimental and computational scientists is dawning. I am an experimental scientist (i.e I could barely use excel let alone do big data analysis when I started my PhD). But I had to learn how to use R and unix to analyse my data because frankly there was no one else. And this learning was something actively encouraged by my supervisor. Every PhD and post doc James has now can do both. It's very important to him. It really should be more important to a lot more supervisors. So here's my experience: I have done two types of sequencing experiments generating data on DNA methylation (Illumina MethylationEPIC Array) AND gene expression data (RNA-Seq) of two non-cancerous cells treated with metformin. What this gives me in a massive amount of raw data (like hundreds and hundreds of gigabytes - the total amount of data generated from my RNA-Seq was roughly 2-3 terabytes...yeah). I took this raw data and ran it through quality control to make sure every piece of information I got is the best quality. I then ran analyses to determine if there are differences between DNA methylation levels or gene expression levels between your untreated and treated samples. For example if I see a consistent, large reduction in DNA methylation in a certain gene that suggests that there is more gene expression at that gene (low methylation = expression, high methylation = no expression). I can actually overlay this with my RNA sequencing data to see if that gene does actually increase expression! It's pretty cool. I mean you have to validate everything you see in your sequencing data with specific experiments but you can find some very interesting things. So what was the catch (besides learning how to make my computer do this stuff)? It takes time. It took me a full year to analyse my DNA methylation data and validate what I found. I have only started analysing my RNA-Seq data and I can tell you I have sat for 7 hours today watching my computer do aligning on my samples (it's only done 5 out of 9). And god forbid I put a comma in the wrong place or didn't add some arbitrary bit of syntax! I honestly spent a full day trying to figure out why the code I had written didn't work after the computer kept spitting out the "how to" file for the function I had used. I had done exactly what the were saying...except I didn't add two dashes, I had only added one. A bloody dash. My take home message is the more information we have the more we know. Seems simple right? The gathering of that data is in the whole pretty easy to be honest and a lot cheaper than it once was (though both of my sequencing experiments cost a total of around £9,000 - £10,000 each to get the data). The challenge is analysing that data. Handling terabytes of data to find even one gene shows a change in expression is a mammoth task. We need more people who can pick up a pipette and run an experiment as well as analyse the results of that experiment. If you are interested in learning any coding language there are lots of resources online as well as courses you can do. My supervisor (James Flanagan) and a fellow bioinformatician (he's a genius - Ed Curry) run a masters in Imperial in cancer informatics. It may seem daunting but if I can learn it so can you. And not just for cancer research or science in general. Knowing how to code could benefit a lot of industries. Bioinformatics! It's the future people! Sure how are you going to programme your robot in the future if you don't know code?? Resources I found useful: www.youtube.com/user/marinstatlectures www.tutorialspoint.com/r/ www.coursera.org/learn/data-scientists-tools www.imperial.ac.uk/study/pg/medicine/cancer-informatics/ 
This year at the Imperial Festival, CRUK wanted to focus on multi-disciplinary research
The second demo was looking at the work the National Physics Laboratory (NPL) are doing on building the "Google Maps of Cancer". The NPL were CRUK Grand Challenge winners giving funding to expand this idea.
The idea is to bring together all the information we have about cancer to go from the whole tumour down to the molecular level. We can use techniques such as Mass Spectrometry to gather information about cancer for example different metabolites produced by cancer compared to normal cells. The demo ask participants to scan the QC code on the blocks. This led to a neat short video about how mass spectrometry worked. At the end you go a result (i.e. the colour the block should be). You turn over the block and leave it. At the end of the day there should be an image from all the blocks turned over. For more information: http://www.npl.co.uk/grandchallenge/
Time done: 30 months
Time left in lab: 9 months Time to write up: 9 months Mood: wildly bouncing from euphoria to depression So I thought I would do a quick update on how my PhD is going since it’s been a while. I am officially ok with my hypothesis! Before Christmas was the hardest my whole PhD has been. I had spent a year analysing and testing my Illumina MethylationEPIC array (which looks at over 850,000 CpG sites across the genome - not that many considering how many there actually are). I found sites that had significant increases and decreases in methylation when I treat with my drug, spent months validating them using pyrosequencing and what did I find? Nothing. It didn’t validate. It was soul crushing. Buuuut I bounced back. Had a massive rethink with James (my supervisor). We thought about what my drug theoretically does and how that could affect epigenetics and I came up with a new, testable hypothesis. We were assuming my drug does a very specific thing that can be reproduced over and over (eg it always causes the same change in DNA methylation in the same place regardless of what cell line I use or how many times I test it). But maybe it’s not so specific but a bit more random, which is totally fine! This new direction also means my Illumina MethylationEPIC array analysis wasn’t a waste of time. Celebrations all around! And it seems to be going well so far. Well except that I have been troubleshooting the same western blot for nearly three months (and beating my head against a wall. Chairs may have nearly been flung through windows). There has been some tears not gonna lie. It’s hard to do the same 2-4 day experiment over and over to prove the result you got in the first place (in bloody January) was correct all along. Science - when you know you’re right but you have to show all the wrong ways to do it to prove you’re right! So what’s the plan now? Well I am going to the American Association for Cancer Research (AACR) Conference 2018 Chicago in April (there will be many vlogs, blogs etc) to present what I’ve found so far! Absolutely terrifying since I’ve only started to believe in my results like....last week. I have about 9 months left of work in the lab before I need to write it all as a thesis. That will consist of learning a few new techniques (RNA-seq, ChIP-PCR) and to get a bit more of an idea of what’s going on and how I can sum this all up in a pretty bow. I’m also getting another masters student to supervise (YAY) which I’m very excited about because I really like the project I wrote and the results regardless of what comes out are gonna be very interesting. She’s starting at the start of May and finishing up in September. I need to write some papers! I apparently have to publish my data (who knew) so the next six months will be writing up what I've done so far in a paper, getting it peer reviewed by people in my group so I can find any holes and plug them before December. I can then sent the actual papers off for real peer review and (hopefully) publication. And that's it. Sounds simple enough but it's taken 7.30am starts working 5-7 days a week to sum up everything I've done since August in a few paragraphs. I've even started drinking coffee in work. I shake violently when I drink coffee. It's not a good idea. I have one last thing to say to anyone thinking of doing a PhD, to people currently doing PhDs or anyone living with a person doing a PhD: To the person thinking of doing a PhD: be prepared. I want to scream at you "DON'T DO IT, IT'LL RUIN YOUR LIFE" but I won't. We need more researchers so we need more PhDs. But I will say be prepared. It is not all rainbows and unicorns. It's hard and it will drain the soul out of you. But then you'll get a result and feel like the smartest person in the world. You will love and hate the 3-4 years of your life doing a PhD. But really...be prepared to hate it a lot more of that time than you think. To the person currently doing their PhD: you are a god damn hero. Keep going. You may feel like you're drowning or that "maybe a masters for this isn't so bad, I should give up". DON'T. You have given your blood, sweat and tears to this thing. You are going to finish. Even if I have to carry you. But seriously. You have got this. You are smarter than you know, you are incredibly dedicated and you will finish. No one knows what doing a PhD feels like until they have done one. No ones knows the soul crushing pain. I know. And I know you have got this. To the person living with a PhD: please be patient. I know you don't understand what they're doing. I know you don't get why a little comma could cause a massive mental breakdown, the consumption of ALL of the ice cream and you getting shouted at but trust me there's a reason. It's hours, weeks, months or years worth of work that your loved one has just watched go down the drain (this specific example is for programmers but is applicable to all types of PhD). Just keep supporting and loving and caring for the lunatic PhD because they need it. And thank you for being there. Your presence means more than you will ever know. PS: I know I haven't been posting a lot. I do apologise but every second of my waking life is going into completing my AACR poster so bear with me. 
PPS: Apparently I have a very neat desk in my office and all other desks should aspire to look like mine......Weird email sent around the office!? So.....don't get too jealous everyone...
Cute animal photos keeping me sane are courtesy of the wonderful Fota Wildlife Park Ranger (Liam) supporting this lunatic PhD (me). 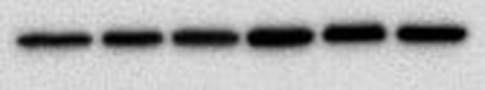 This is going to be a long and technical one sorry. This month I want to talk about an experiment I've done pretty much every day for the last three months (plus continuously over the last 2.5 years) - Western Blot. Western blots are used to visualise and identify individual proteins. In this technique you have a solution (called a lysate) filled with the majority of proteins from cells. This technique separates these proteins out by size and allows you to stain for specific proteins you want (more on this later). This is the standard way to visualise proteins but it is very subjective and is tricky to perfect (I hope you will appreciate why after reading this). The proteins are visualised by staining them with antibodies specific to that protein and then using a chemical reaction to produce light which can be picked up by a machine. The resulting image is of black bands. The intensity of the black band determines the approximate level of protein present. There are a few steps to Western Blotting which I will go through. The whole technique can take 1-3 days depending on what proteins you are looking for and what you choose to do in each step. Steps to Western Blotting:
A little bit of terminology before we begin:
STEP 1: Preparation Reagents and Buffers Before you begin a western you need to make sure you have all your reagents prepared. I will NOT be going through this, all I will say is you need reagents for
Protein Preparation: When you extract your protein from your cells you end up with a protein lysate. This contains the majority of the proteins in your cells. You don't know what the concentration of the total protein is but you are sure that each sample doesn't contain the same concentration. This is due to how well the extraction went in each individual sample but also how many cells there is to extract protein from. There are a few ways of finding out the concentration of your protein which I won't go through (I use the Bradford Assay from BioRad). What you do need to know is that for each sample you need to make up a solution of protein, water and loading buffer to load onto your gel. You can use as much or as little protein as you want, I typically use 10-15ug per well. I load 20ul per well so I need:
There are many types of loading buffer, I use Laemmli buffer. The loading buffer has a couple of functions. You are using an electric current to make the proteins travel through the gel. Proteins are all different charges so the buffer makes sure all the proteins have the same charge (negative) - this is done by a chemical called SDS. Proteins are globular structures. These don't run though a gel very well so you need to unravel the proteins into single strands (called denaturation). The buffer will start this process - a chemical called β-mercaptoethanol breaks disulphide bonds. The loading buffer also has a dye in it. This is called the "loading front" which runs faster than your proteins. This allows you to track how far your proteins have travelled through the gel (image later).
A quick note - usually the protein samples you have prepared will be blue once you add the loading buffer and boil it. This is because the dye in the loading buffer is blue. However, the protein I will show in this post will be yellow. This is because I extracted the proteins using acid and the acid in the lysate turns the buffer yellow. Once the gel starts running the dye front goes back to blue.
Gels are made up of:
The acrylamide will set into a gel which has pores in it. The protein will travel through these pores. The percentage of acrylamide used determines how big these pores are. Large proteins run slowly through a gel whereas smaller proteins will run very quickly through the gel. A lower percentage gel (e.g. 7%) will have large pores, allowing larger proteins to run faster through the gel. However you are likely to lose all the smallest proteins. A higher percentage gel (e.g. 12.5%) will have very small pores which slow down the small proteins, allowing you to visualise them easier. However all your largest proteins might not run as they can't past through the pores. There are two types of gels used in gel electrophoresis - a stacking gel and a resolving gel. They're pretty much made up of the same ingredients (a stacking gel is slightly more acidic pH 6.8).
Steps to making a gel:
I have made up a 12.5% gel for this example to run histone proteins which are approx 15-17kDa in size. I will also stain for a loading control, beta actin, which is 45kDa in size. STEP 2: Loading and running SDS-PAGE Gel Electrophoresis The next step is to load your prepared protein into the wells in your stacking gel and running an electric current through the gel to allow the proteins to travel from negative to positive. The gel(s) are clamped into an electrode chamber and placed in a running tank. The centre is filled with running buffer. The running buffer allows an efficient flow of electrical current to pass through the gels. It also prevents the gels from drying out which you do not want.
STEP 3: Transferring proteins from gel to PVDF membrane Once the gel has run enough you can stop it. This depends on how long you want to wait for and what proteins you're looking for. My proteins are very small (17kDa) so I can stop my gel once I see the 17kDa ladder band is far enough from the loading front. The next step is to transfer the proteins from the gel onto a membrane. You cannot stain for your proteins on the gel as it is far to delicate and not permeable - the protein only runs through the gel because we created the wells. The membrane is more permeable and allows you to stain for proteins. There are a couple of (messy and time consuming) ways to transfer proteins but I prefer to use the iBlot dry technique. This basically transfers the proteins using a machine in 5 minutes (compared to 1-2 hours). The transfer stack consists of:
Again an electric current is used to push the proteins through the gel into the membrane. However the voltage is very low. As we know, small proteins move very quickly. The low voltage prevents the small proteins from passing straight through the membrane. So how does it work:
 Image of how you set up before transfer
After the transfer is complete you need to be VERY fast. You have to quickly remove the stack from the machine, get the membrane out of the stack and place it in TBST. The membrane CANNOT dry out or else all is lost. You can check your transfer at this stage using Ponceau red, which is a red dye that shows up all the wells and bands of proteins but I did not do that today. STEP 4: Blocking The next step is to block any unspecific binding. The antibodies are usually polyclonal - meaning they can bind to other proteins that aren't your protein of interest. To prevent this and also to get a nice clean image at the end, you block unspecific binding. You can block using either of these two reagents:
The reagent you are using depends on the antibodies and proteins you are looking for. I block in 5% BSA/TBST/NaN3. Before I block I cut my membrane so that I have the two individual gels again. You don't have to do this but I like to. At this point I just cut the excess off using the ladders as a guide. I then mark with a pen on the ladder what gel it is. The membranes are blocked for 1 hour at room temperature on a rocker. This is now when we can call out membrane a blot. After blocking you can cut your blots to stain for multiple proteins. In this case I cut at 36kDa band (blue band above the bottom red band on the ladder) to stain for my histone proteins and also my beta actin loading control.
STEP 5: Antibody Staining of Proteins This technique as I said uses antibodies which bind to specific proteins and you can them visualise these antibodies using a chemical reaction. The figure below gives a rough idea of how the technique works.
The antibodies are created in different animal models (please don't ask how cause I have no idea). So if your primary antibody is developed in a rabbit then the secondary antibody will be goat anti-rabbit (i.e an antibody developed in a goat to bind specifically to rabbit-developed antibodies). There are some human antibodies but they're incredibly expensive. The primary antibodies are made up in the buffer recommended by the manufacturer usually 5% BSA/TBST/NaN3. The recommended concentration is usually given by the manufacturer - typically 1:1000 or 1:2000 (i.e. 1ul antibody in 1ml solvent). The secondary antibodies are made up in usually either 5% BSA/TBST or 5% Milk/TBST. The BSA doesn't contain NaN3 in the secondary because sodium azide (NaN3) inhibits the secondary antibody. The concentration of the secondary depends on how abundant the protein is and how long you develop it for. For example beta actin, which makes up part of the cytoskeleton of your cells, is an incredibly abundant protein so the secondary concentration is 1:50,000 BUT another protein I look at p-AMPK is not so abundant and the secondary concentration could be as low as 1:1000. 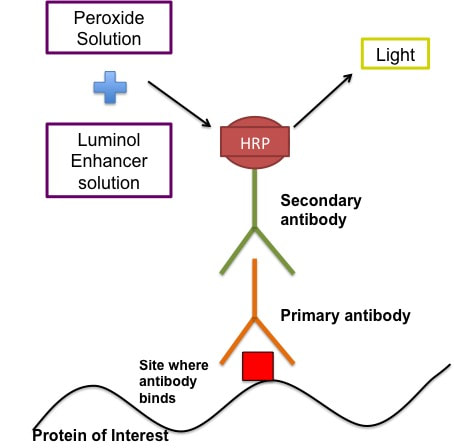 Diagram of antibody staining of proteins Part 1: Primary antibody incubation and washing
NOTE: for beta actin, you do a 1 hour incubation at room temperature Part 2: Secondary antibody incubation and washing For the secondary antibody you can do this at room temperature for 30 minutes - 1 hour, again on a roller. I make up most of my secondaries in 5% Milk/TBST, except p-AMPK. The casein in the milk removes phosphorylation from proteins so I would lose my p from the p-AMPK if I did the secondary in milk. So for p-AMPK and all phosphorylation proteins I use 5% BSA/TBST. Again after incubation in the secondary, the blot(s) need to be washed at least 6x5 minutes to remove excess secondary antibody. The washing part of the procedure is honestly the most time consuming and tedious. It's a lot of back and forth to and from my office desk to the rocker. I usually try to do something else in the lab to not waste the time but sometimes it can't be avoided. STEP 6: Developing The final step (thank god) is to develop your blot. This step adds the peroxide and luminol enhancer solutions (collectively known as ECL solutions) to the membranes. You then take the membranes to a chemiluminescence machine which will detect the light emitted and give you an image in return. Steps:
 Image of the chemiluminescence machine You end up with a picture which has black bands. As I said at the start the intensity of the bands indicates how much protein is present - very dark means a lot, very light means very little and nothing means nothing. 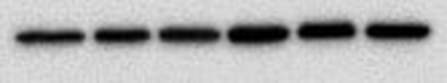 Image of a beta actin western blot for MCF10A (lane 1-3) and MCF12A (lane 4-6). The image above shows the loading control (beta actin) for a blot I have done for two cell lines, MCF10A (lane 1-3) and MCF12A (lane 4-6). The bands are very dark indicating that the protein is very abundant. What's important is the bands are almost the exact same size and intensity as each other (different in the two cell lines) indicating that my loading was very even (wahey). Once the blot is developed you can keep it or you can throw it away. If you keep it and want to stain for another protein or the same protein but with a different secondary concentration you need to strip the blot (i.e. remove all antibodies bound to proteins), re-block using the same blocking buffer and re-incubate with the primary and secondary antibodies. A blot should only really be stripped twice. This will add another day onto your experiment (at least). And that is it. That is roughly how a western blot works. The reason why western blots are THE most hated laboratory experiment is because they rarely go the way you want them to, there is always a difference each time you do it and it takes FOREVER. If a western goes wrong most of the time you have no idea why. It can be anything from one specific buffer which was made up wrong to the wrong gel percentage, the wrong transfer time or transfer voltage etc. Basically everything you did could have an issue associated with it. Trust me I did troubleshooting for TWO MONTHS on one antibody. It eats away at your soul. Western blotting everyone. 
Image from https://www.alcoholconcern.org.uk/dry-january
Happy New Year!!
January is the month where New Year's resolutions are made...and broken. We all want to get fit, eat healthier and improve our wellbeing. A brilliant campaign that combines fundraising, awareness and improving your health is Dry January. Many charities encourage people to stop drinking alcohol for one month in order to raise awareness about a number of different diseases linked to alcohol consumption.
Since I am a cancer researcher (in case you haven't noticed) I am going to talk to you about how alcohol can increase your risk of cancer and why moderation is always key! 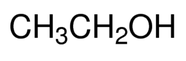 Ethanol chemical formula Ethanol chemical formula
What is alcohol?
What do I mean by alcohol increasing risk and does this change depending on the type of drink? Are light beers better for you compared to vodka? So what is alcohol? Well interestingly alcohols are not the beverages we think of but a group of chemicals all sharing similar properties (they all contain CH3OH). So methanol, ethanol and propanol are all members of the same alcohol family. When we talk about the beverage "alcohol" we are talking about ethanol. Now 100% ethanol is extremely toxic and flammable. You should NEVER drink 100% ethanol, the majority of alcoholic beverages range between 2% - 40%. For reference, I use 70% ethanol to clean my TC hood to remove cells and microbes to maintain the sterility of TC. Actually in the lab we use ethanol for a number of things including "washing" DNA. The way we extract DNA means there are chemicals mixed up with the DNA (like isopropanol). We want our DNA to be as pure as possible so to get rid of those chemicals we "wash" the DNA (in the form of a pellet) with 70-80% ethanol and allow the residual ethanol to evaporate off, leaving us some (hopefully) pure DNA.
Alcohol increases cancer risk and incidence of cancer:
Alcohol has been shown to increase the risk and actually cause seven different cancer types (shown below), but has been implicated in more cancer types. In fact when we talk about the major risk factors for any cancers the top three is always age, BMI and alcohol consumption. The impact of alcohol on cancer depends on the cancer type, for example studies have shown that if a woman drinks 1.5 units a day her risk of breast cancer increases by 5%. Now that doesn't seem like a lot but that percentage increases the more your drink. The risk for cancers such as liver or mouth cancer have a far higher increased risk of cancer with just 1.5 units a day (liver cancer is 16%). 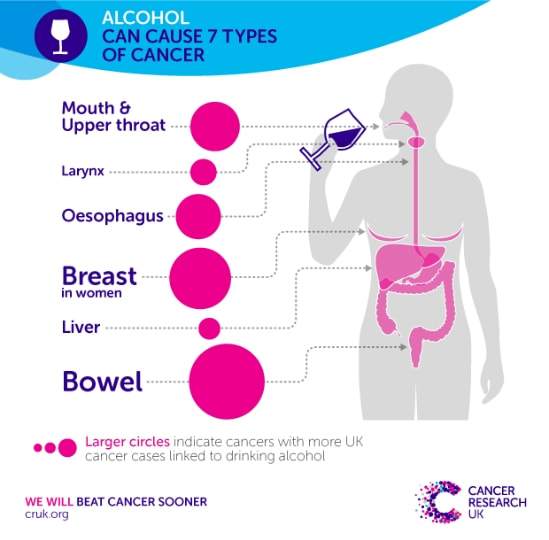
Image from: http://www.cancerresearchuk.org/about-cancer/causes-of-cancer/alcohol-and-cancer/how-alcohol-causes-cancer
How does Alcohol cause Cancer?
Alcohol is classified as a carcinogen (since 1988 by IARC). Carcinogens are substances that cause damage to any part of a living cell. Alcohol is metabolised in the cell to acetaldehyde and this has the carcinogenic properties. There are a number of ways alcohol can cause cancer:
As I've talked about before, cancer in general is an accumulation of small changes to normal biological pathways such as DNA damage, changes in hormone levels, changes in nutrient levels and over activation of certain pathways. This all contributes to the cells becoming cancerous. Alcohol can generate and help increase these pathways, making it easier for cells to become cancerous. In fact alcohol actually increase risk further when combined with smoking (something people commonly do when they drink). This is because cigarettes contain other carcinogens which also damage DNA etc. So you're getting a double hit of damage at once.
What is the recommended intake of alcohol?
We hear a lot about the "recommended units of alcohol" per person, per week. But what is that? Well first of all a "unit" is a measurement which depends on percentage alcohol and volume of the liquid. So the unit depends on the type of alcohol you're drinking, for example 1 unit of spirits is 25ml volume of 40% alcohol but 1 unit of beer is about 225ml of 4% alcohol. The new guidelines (released a couple of years ago) recommend that men and women should drink no more than 14 units of alcohol per week BUT stipulates this must be spread over at least THREE DAYS. Meaning for example you can have 6 glasses of wine a week but you can only have four glasses max in one day. This is to try reduce binge drinking. Binge drinking is defined by the Office of National Statistics as over 8 units of alcohol for men in a single sitting (about 3 pints) or 6 units for women in a single sitting (about 2.5 glasses of wine) or drinking a large amount of alcohol in a short period of time. 
Image from: https://www.drinkaware.co.uk/alcohol-facts/alcoholic-drinks-units/alcohol-limits-unit-guidelines/
The take home message:
It's all about moderation! If you enjoy a glass of wine in the evening that's fine but maybe one day of the week you don't have that glass of wine! It's important to be aware of the volume you are drinking. Pints are easier because pint glasses are standardised but wine glasses can come in different shapes and sizes. How do you know you're drinking 175ml? It's also about feeling better and improving your overall health. Alcohol is not just linked to cancer but other diseases like cardiovascular disease. Alcohol can also contribute to weight gain, especially high sugar drinks like larger, cider and/or wine. Ask anyone who has done Dry January before and they will tell you how good they feel, even after just a few weeks. I'm not preaching here. I'm not saying GIVE UP ALCOHOL NOW! I enjoy a drink but I don't drink very often as a choice. What I am suggesting that maybe reducing you alcohol intake could improve your health and overall lifestyle for the better. January is the month to try it out while you're surrounded by other "designated drivers", see how it benefits your health (and your pocket).
If you want to become a Dryathlete or just find out more about the campaigns you can visit:
www.cancerresearchuk.org/support-us/do-your-own-fundraising/dryathlon https://www.alcoholconcern.org.uk/dry-january www.drinkaware.co.uk/alcohol-facts/alcoholic-drinks-units/alcohol-limits-unit-guidelines/ www.cancer.gov/about-cancer/causes-prevention/risk/alcohol/alcohol-fact-sheet 
November is a month of awareness for a few cancers. I will discuss each briefly but you can find more detail in the links given below.
I'm going to start with the worst here, pancreatic cancer. Pancreatic Cancer: Pancreatic cancer is the 11th most common cancer in the UK accounting for about 10,000 cases each year. The incidence has been steadily increasing over the last 10 years, more so in women than in men. It only accounts for about 5% of cancer-related deaths each year but here's the kicker...less than 1% of pancreatic patients will survive 10 years or more. The five-year survival rate? 3%. And finally only 20% of patients will survive their first year. That is a loss on average of around 8,000 lives a year just down to one cancer type. So why is this cancer type so bad. There are a number of reasons, the first being your pancreas is pretty important for your survival. Your pancreas does two vital things, it produces enzymes which help you digest your food and it produces hormones to regulate your blood glucose levels. Hormones like insulin. The second reason pancreatic cancer is so hard to treat is its location. Your pancreas is embedded deep in your abdomen between your stomach and your spine. The third reason is your pancreas doesn't regenerate and it's very difficult to replace with a donor. And finally and possibly most importantly for cancer treatment, pancreatic cancer usually presents at a very late stage meaning very little can be done. Symptoms: Again with a lot of cancers the symptoms are vague and can include:
Type of pancreatic cancer:
Diagnosis and treatment: Diagnosis of pancreatic cancer is mainly through examination of the pancreas using an ultrasound or CT scan accompanied by blood tests and a biopsy. The majority of patients are diagnosed at stage IV, which is why the survival rate is so low. The treatments for pancreatic cancer are the standard: surgery, chemotherapy and/or radiotherapy. Unfortunately lack of response to treatment and recurrence is high. There are many ongoing clinical trails to find new targeted therapies for pancreatic cancer. Screening and prevention: Currently there are no screening programmes for pancreatic cancer and early diagnostic tools like CA19-9 are unreliable as a proportion of pancreatic cancers don't release this marker. This is a terrible disease with a shocking survival rate and it is only getting worse. Pancreatic cancer research is one of the most underfunded research areas today. But with more awareness hopefully charities like Pancreatic Cancer UK can raise more funds for vital research.
To keep with the theme of vastly underfunded research areas I will next talk about
Lung Cancer: You would expect through all the campaigns about quitting smoking etc. that lung cancer would be one of the most funded cancer research areas but it is sadly not. Lung cancer is the third most commonly diagnosed cancer in the UK accounting for about 46,000 new cases a year. But just like pancreatic cancer, the 10-year survival rate is only 5%. The sad thing is CRUK estimates about 89% of lung cancer cases could be prevented. I won't lie to you we all know the biggest risk factor for lung cancer is smoking. We've seen the ads and the photos on cigarette packets. The statistic here to remember is that while only 10% of smokers will get lung cancer a whopping 86% of lung cancer patients are smokers, former smokers or passive smokers. Symptoms of Lung Cancer:
Types of lung cancer: The "types" of lung cancer are grouped together because of the size of cell the cancer originated in but they are not all the same cancer.
Diagnosis and treatment: Usually once you have presented with symptoms which indicate lung cancer a patient will receive a chest x-ray and/or bronchoscopy accompanied by a biopsy. It must be noted that treatment for the two cancer types are different, depending on where it is and the stage. But the standard treatments for lung cancer is standard: surgery (which could be as little as removing the tumour to as much as removing a lung), chemotherapy, radiotherapy, photodynamic therapy and ablation. There are targeted therapies available for advanced lung cancer patients which target pathways such as EGFR (erlotinib) or tyrosine kinases (genfitinib). Screening and prevention: While there is no national screening taking place for lung cancer the best way to prevent it is to quit smoking. Anti-smoking campaigns are one of the most successful awareness campaigns out there. And the big question you've been burning to ask - why is lung cancer so underfunded? Sadly I have no answer for you. The funding process is a difficult and complex system which I know very little about. The good news is that CRUK at least have pledged to put more funding into these poorly researched cancers like Lung and Pancreatic.
And finally let's talk about Movember. Now November isn't the month for awareness on testicular cancer or prostate cancer so I won't be talking about these. However it has become a month where awareness and funds are raised for all men's cancers. And awareness to me is more important that a single penny. I have spoken on this before but just to hit it home once again, prevention is better than cure. Not all cancers can be prevented I'm afraid but if we can reduce the numbers of people being diagnosed we can reduce the numbers who lose their lives.
The fact the Movember has come about is particularly important. The most funded cancer research area to date is breast cancer. And it is thought that part of the reason for this is women. Women who had/have breast cancer or women who have lost a family member or friend to breast cancer. These women are the driving force behind every awareness campaign, behind new drugs being made available to patients and behind vastly successful national screening. To me it seems women are, interestingly, the major focus of cancer awareness campaigns because they are usually the driving force behind them. I'm not going to go into gender biases about how "women talk more about their problems than men" because if cancer screening has taught me anything its not true (take a look at bowel cancer screening statistics). But what I am saying is that men as a whole (sorry to put it like that) have less opportunities to avail of screening services and may not know what they are really supposed to be looking out for. Which is why Movember is so brilliant. It encourages men to get involved and to talk about male cancers. It puts male-specific cancers in focus while allowing men to actively participate by growing beards (super manly). I like to highlight charities and companies that inspire me and to me show genius and innovative ways of raising cancer awareness and funds. And the underwear company OddBalls encompasses this completely. The company, started in 2014, designs and creates underwear (not just for men but for women as well). And with your new boxers you also receive a handy guide to checking yourself for testicular cancer. They have become so popular they now design underwear for rugby clubs including The Welsh International Team AND have set up their own charitable foundation. If you want to know more or maybe by a pair for yourself, check out: www.myoddballs.com/ They also do socks, bobble hats, rugby jerseys and more!
If you have any questions please don't hesitate to contact me.
For more information visit: www.pancreaticcancer.org.uk/ www.cancerresearchuk.org/about-cancer/pancreatic-cancer?ds_kids=p3584232188&adc=cpc&gclid=EAIaIQobChMImP__2rjG2AIVyrftCh0JBwOcEAAYAiAAEgLai_D_BwE&dclid=CMSp4ty4xtgCFVBuGwodM9sKyg www.cancerresearchuk.org/about-cancer/lung-cancer www.blf.org.uk/?gclid=EAIaIQobChMIqZy7osLG2AIVzbztCh2LnAvCEAAYASAAEgIt3fD_BwE uk.movember.com/ 
October is breast cancer awareness month. I want to tell you in this post some information about breast cancer symptoms, diagnosis and treatments. I have worked in various areas of breast cancer research in the last 5 years, most notably my PhD is focused on understanding diabetic drugs in order to prevent breast cancer. It's an area close to my heart.
Breast cancer is the most commonly diagnosed cancer in the UK. About 55,000 new cases are diagnosed every year and 7% of all cancer-related deaths can be attributed to breast cancer. There is some good news and bad news about breast cancer survival. The overall 5-year survival is around 86% but when you break this down by stage of the cancer type, this value falls dramatically for those with higher stage cancers. 5-year survival for women with stage IV cancer is approximately 15%. The survival rate has doubled in the last 40 years due to better preventative measures, better diagnosis and better therapies. 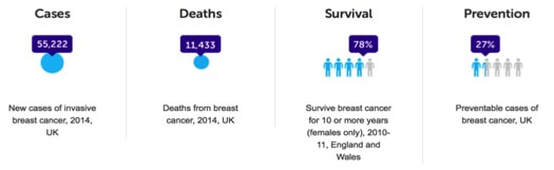
Image 1: Breast cancer statistics. Image from http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/breast-cancer
Breast cancer types:
I've already spoken a bit on the different cancer types and how tumours can be classed by their origin but also molecular differences ("What the Hell? - Cancer Part 2). 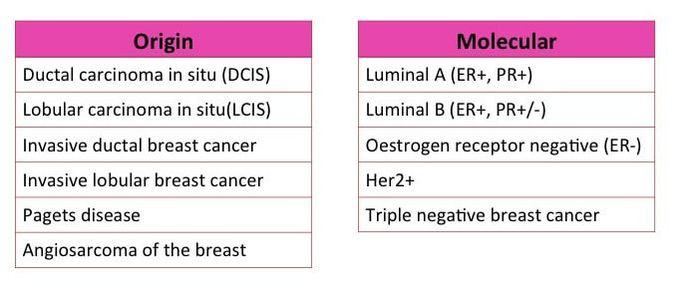
Image 2: Types of breast cancer
I've also talked before about risk factors, which are lifestyle or genetic factors that can increase or decrease the likelihood of developing a certain disease. There are a number of risk factors associated with breast cancer (some are in image 3).
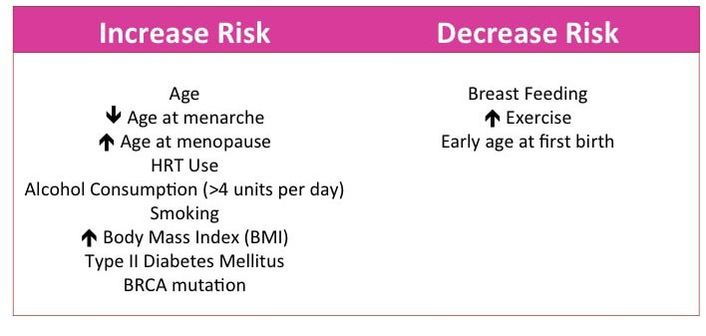
Image 3: Risk factors associated with breast cancer
Symptoms:
Most women know the symptoms of breast cancer:
Diagnosis: There are a number of ways a breast tumour is identified:
Treatment: Treatment depends on the stage/grade of your tumour, the molecular subtype and the willingness of the patient. There are the standard treatment practices (1-3) but also more personalised therapies (4-5)
Prevention: Prevention is key. You will hear my harp on about prevention is the cure to cancer but really it's one of the key areas we as individuals can look after for our own health. In fact it’s estimated that about 27% of all breast cancers could be prevented and there are a number of prevention techniques you can adopt
All rights to this video belong to Coppafeel!.org
And let's not forget the men here! Men can suffer with breast cancer. While only about 400 men are diagnosed each year that is still a significant number. Male breast cancer is quite similar to female breast cancer in terms of risks (though an additional risk factor is Klinefelter's syndrome), diagnosis, symptoms and treatments.
Finally I would like to talk to you about a charity which inspires me to keep doing the work I do. This charity was set up by a young woman who was diagnosed with breast cancer in her early 20's. Instead of wallowing in her diagnosis of metastatic breast cancer she decided to set up a charity to better educate young women to CHECK YOUR BREASTS! That charity was set up over 8 years ago and despite multiple metastatic tumours Kris is still alive and more vibrant than ever. This charity is called Coppafeel! (a genius name). If you have time please check out their website https://coppafeel.org/ and check out Kris's story (https://coppafeel.org/our-charity/kris-story/). A truly inspirational woman backed by the most incredible fundraisers and supporters. I haven't had the chance to work with coppafeel but it is one of the charities I would most like to get involved in.
For more information on any of the areas I've discussed visit:
about-cancer.cancerresearchuk.org/about-cancer/breast-cancer www.breastcancernow.org 
The staff and the organisers of the event were brilliant and so accommodating. They have this wonderful sort of art installation where the public/researchers at the Crick ask "What...the one thing [they] would like to know about the human body, how disease develops or how they might be treated?" I found one I thought was very insightful from a 17 year old, "Do atoms have motives?".

Day 1:
The first day opened with two key note speeches from two extraordinary researchers. The first speaker, Dr Lewis Cantley, discovered the PI3K signalling pathway (which is a big deal trust me, this is one of the biggest signalling pathways there is and it is incredibly important in cancer biology). He actually spoke about something very close to my own research. Insulin resistance (hyperinsulinemia) is a risk factor for many types of cancers. The liver, skeletal muscle etc. become resistant to insulin signalling, leading to a build up of insulin in the blood. BUT cancer cells are highly sensitive to insulin, meaning this high blood insulin environment is perfect for the cancer to grow in. Insulin itself activates pathways to make cells grow and divide. PI3K is part of the insulin signalling pathway. Dr Cantley talked about how using PI3K inhibitors (which are already being used but can become ineffective) in combination with a modified diet could help enhance PI3K inhibition for cancer treatment. And funnily enough this PI3K and metabolism subject came up a lot throughout the two and a half days, not just in the "Metabolism" session. The second speaker was Dr. Lisa Coussens, who is a big name in cancer immune biology. Dr. Coussens spoke about the immune cells in the tumour microenvironment and how this can contribute to inflammation around tumour cells. She also spoke about how immune cell infiltration into tissue can contribute to tumour development and metastasis. Understanding the different immune cell infiltrates will help is understanding tumour development and also potential treatment.
Day 2:
The second day was long, very long (11 hours from arriving to leaving). The day was packed with talks and poster presentations. I won't go through every single speaker's work in detail but I will highlight some areas I found particularly interesting. My main reason for attending this conference was the session on metabolism. I have five pages of small scribbled notes in my notebook from the five talks compared to one or two for the other sessions. Not the other sessions weren't interesting. Unfortunately I can't really discuss a huge amount of what was talked about because most of the data is unpublished and you have a professional courtesy not to disclose the information. Session 1: Metabolsim This session really focused on how understanding metabolic processes, the metabolites that are used in these processes and the metabolites derived from those processes could help in not only understanding how tumours are formed but also how was can target these tumours as individual treatments themselves or in fact enhance current treatments. As I've said before it's been shown that different diets can enhance the effect of anti-cancer drugs. Session 2: Tumour Microenvironment I've talked about the tumour microenvironment (TME) before in my "What the hell? - Cancer Part 1" post. The TME is made up of a number of different cell types (immune cells, blood vessel cells etc.) which all contribute to the development, growth and spread of cancer. In this session we discussed how we can understand the interaction between the cells in the TME and cancer cells. Mainly this session focused on infiltrating immune cells (immunology is a hot topic now so it came up in pretty much every talk at some point....) but there was one talk which was very interesting about identifying mutational signatures in breast cancer that can identify the type of cancer it is, can put together cancers that may not look the same but have similar mutations and can tell you about potential treatment pathways (Serena Nik-Zainal).
Day 3:
Session 3: Tumour Immune system interactions As I said immunology in cancer is a hot topic in recent years. This session focused on targeting immune cells to kill cancer. Immune cells can be pro tumour and anti-tumour depending on the signals it received. Cancer cells can send out signals that activate immune cells that dampen down the immune response while also pretending they're normal cells to immune cells that would kill them. Immune cell infiltration into the tumour can happen at different stages of tumour development and the context (I learnt) is pretty important for choosing what cells to inhibit to kill cancer cells. Interestingly the metabolism/immunology sessions combined for a talk by Luke O'Neill (a lecturer in Trinity College Dublin). While by far the most entertaining lecture he also made an interesting point about how different metabolites can be pro-inflammation or anti-inflammation. While he doesn't focus on cancer, he made the interesting point that targeting immune cells could be again achieved or enhanced by looking at the metabolism of the cell you're targeting. Session 4: Tumour heterogeneity and evolution The final session was a mixture in terms of biology and statistics. We had talks about cancer evolution, specifically the evolution of drug resistance (drug resistance is a big problem in all cancers) and heterogeneity of cancers. If we can understand how drug resistance occurs we could target cancer cells better so it doesn't happen. A lot of that has to with the fact that tumours can have multiple cancer cell sub-groups which have different mutations (heterogeneity). The statistics talk was interesting but I was very much out of my depth. What really fascinated me and kind of scared me was the talk on brain tumours. Basically brain tumours act like mini-brains. They form cell-to-cell connections through micro-tubules. This tubes allow the cell mass to become more resilient to therapy (e.g. radiation) and also allow repair of damaged parts of the cell network (which brains do not do). Frank Winkler showed videos of the brain tumour cells and even compared them to normal brain cell connections. While scary to know, this is brilliant for brain tumour biology and treatment. Recurrence is common in brain tumours and these tubules could help give an answer as to why. And they could be something that can be targeted. More work needs to be done but that talk was brilliant.
Posters:
I first want to explain what a poster session is for those who don't know. Basically you submit a short description of your work and if you're chosen you create a poster (A2) to demonstrate what your work. The more figures the better. You then have an opportunity to stand beside the poster and chat to any interested person who comes along. I want to thank the people who happily stood and answered my weird questions about their work. You're very patient humans. I want to say I went and talked to everyone about their posters (there were 74 by the way) but I hunted out the epigenetics posters and ones that really interested me. I chatted with Tim Fenton from UCL, James Heward from Barts and Neil Slaven from Imperial. I can't really tell you what we talked about because their work is unpublished but needless to say it was very interesting.
All I all it was a really interesting conference, if not a a small bit tiring. My only problem with the conference as a whole was the very small representation from the area of epigenetics. Some speakers mentioned epigenetics (one even had a little data) and there were a few posters on the subject but it was distinctly lacking. I know that's the area I'm in so I am very biased and obviously it was hard enough to fit all the talks they had already into the two and a half day schedule, but it was missed. It may not be the area that solves all the problems (my PhD title will of course be "I cured cancer with epigenetics, everyone can go home now") but it provides a key piece in the puzzle. It's something I hope the organisers will consider for the next Crick International Cancer Conference.
Other than that the 1st Crick International Cancer Conference was worth attending and I learnt some interesting (and terrifying) stuff.
You may (or may not) have noticed that my blog has been all but quiet and desolate the last three months. This is completely my fault and an unconscious act of neglect. There are two main reasons why I have been so absent and hopefully I can give you many excuses to explain these reasons while also updating you on my progress.
The first reason is work. In mid-August I had what's known here as a Late Stage Review (LSR). This is something all Imperial PhD students are required to do in order to complete their PhD. The aim is to present your results and future plans after around 2 years of doing your PhD. You make up a report and do a presentation for two examiners. They judge your work and your ability to complete your PhD and sign you off. Usually they don't fail people (there is always the possibility) and the idea is for them to give you ideas for finishing your thesis and focusing your work. Obviously you want to present as much good data as possible. Hence the absence beforehand. I spent all my time writing the report and generating as much data as I could. All in all the LSR went fine. My work is satisfactory enough to continue. However I am not going to lie I was disappointed afterwards. I thought going in that they would help me narrow down and focus on the more important aspects of my project but instead I was told to "expand more to focus more". I realistically have 12 months left in the lab. This was not what I wanted to hear. I need to add more work to quite frankly an already daunting amount of work. Following from that I wanted to finish up at least one part of my project (validating my significant probes found from the DNA methylation array in another format i.e pyrosequencing). And this you will be happy to know showed...........nothing. Bupkis. Nada. I have negative results. While this is not an epic failure (as my brain wants me to believe) nor is it a triumph. Essentially what this means is I need to go back to my plan and look at the question a different way. And this is when I appreciated the LSR. Yes I needed to do more work but actually it gave me an opportunity to approach my project at a slightly different angle and who knows maybe something will work. I am now in the stages of planning and implementing that work. And who knows maybe in a few months time I will have something positive to talk about. The other reason I have been absent is I went on a 17 day holiday. I travelled to Rome, Sicily, Dublin and Cork. It was amazing and relaxing. And I didn't have to worry about my project or stress about what I needed to do next. I even slept the whole way through a night for the first time in, well, years. It was incredible and for anyone looking for their next holiday destination I highly recommend Sicily. Get a car like we did, drive around and soak it all in. Mountains, crystal clear water, food. A must really. And finally on a slightly different note I am taking up some teaching! An ex-member of our lab, Kirsty, is part of a team which has created a brand new innovative BSc undergraduate programme in Imperial. The course focuses on interactive learning and face-to-face teaching. It will be one of the first courses of its kind and hopefully not the last. Part of this course is to teach laboratory skills, called lab pods. I will basically be a part of a team of PhD students in these lab pods who will help guide students through techniques like cell culture and western blotting. Guide being the operative word. Unlike traditional teaching labs where I would show them what to do and answer all of their questions for them, this lab aims to make scientists. So I am really there to watch and provide last resort help. The idea is that the students will get a protocol, read through it and do the experiment. If it goes wrong then they have to figure out why. Exactly like anyone working in a lab would. Besides having a master student, I haven't really taught before so this is a new and exciting experience! Terrifying (especially when you realise you're about 10 years older than they are) but exciting. And that's what I've been doing. hopefully I will be a bit more dedicated to the blog and I can keep you updated on my work, my teaching experience and also complete a few posts (such as my "What the Hell? - Cancer" series). Btw I am not quitting this PhD any time soon in case you were worried. |
AuthorMy name is Caitriona and I am a PhD student at Imperial College London, UK. Categories
All
|




















